
Profile

Alexander Grishaev
Dr. Alexander Grishaev’s laboratory focuses on integrative structural biology and deriving biomolecular structures by combining experimental data from several complementary, biophysical techniques within a computational framework that optimally restrains the conformation space. The lab’s primary experimental approaches are solution X-ray and neutron scattering, and nuclear magnetic resonance (NMR) spectroscopy. They aim to design methodologies for maximizing the information content and fidelity of interpretation of the observables attainable by these techniques. An important part of the Grishaev lab’s work is mining information in structural databases to improve force fields for protein/RNA/DNA structure refinement. The group focuses on systems characterized by a low density of experimental restraints, such as flexibly linked multi-domain or disordered proteins, amyloid fibrils, protein complexes, and RNA constructs.
CURRENT RESEARCH
Using wide-angle solution X-ray scattering to define biomolecular structure and dynamics

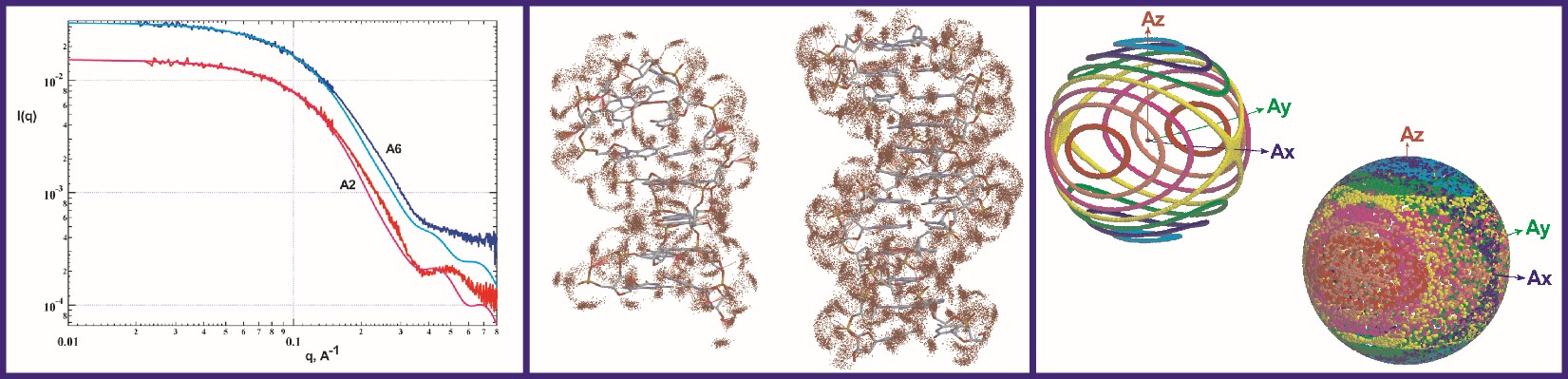
The Grishaev lab develops methods for improving both the speed and the accuracy of modeling solution X-ray scattering data from atomic coordinates to facilitate their integration into structure refinement of intrinsically disordered and flexibly linked multi-domain proteins, RNA, DNA, and lipid or detergent-based nanoparticles. The lab’s ultimate goal is to determine the structures of proteins and RNA as multi-state conformational ensembles, which they view as the next frontier of structural biology. Model improvements are underpinned by high-fidelity molecular dynamics simulations in explicit solvent and structural database analysis, allowing formulation of detailed, experimentally verifiable, multi-dimensional hydration maps and motional models of biomolecules. Infrastructure support for this work comes from state-of-the-art facilities including in-house small- and wide-angle X-ray scattering (SAXS/WAXS) instrumentation, synchrotron beamlines for both non-resonant and anomalous scattering experiments, local neutron scattering facilities at NIST, and a high-performance computing cluster at IBBR. The Grishaev group’s strategy of jointly using wide-angle solution scattering data, NMR, and advanced simulation techniques aims to maximize the density of the experimental observables while limiting the expansion of the number of degrees of freedom to minimize data over-fitting. The group also develops novel scattering approaches -- including high-contrast heavy atoms labels and anisotropic alignment -- to increase the information content of scattering data.
Improving the accuracy of NMR-determined biomolecular structures via better force fields and advanced simulation techniques
NMR is an important component of the lab’s workflow and their efforts include increasing the impact of NMR restraints, such as residual dipolar couplings and chemical shifts. They develop both novel empirical energy terms and codes for better use of NMR and SAXS data, with applications to structure refinement packages, such as Xplor-NIH, CNS, and Amber. They also develop and apply metrics for both empirical protein and RNA structure validation and experimental cross-validation as sensitive and objective gauges of the coordinates’ accuracy.
.
Publications
- Conformational dynamics and multimodal interaction of Paxillin with the focal adhesion targeting domain.
- Critical Assessment of RNA and DNA Structure Predictions via Artificial Intelligence: The Imitation Game.
- Structure and dynamics of monoclonal antibody domains determined using spins, scattering, and simulations.
- Conformational dynamics and multi-modal interaction of Paxillin with the Focal Adhesion Targeting Domain.
- Extracting Orientation and Distance-Dependent Interaction Potentials between Proteins in Solutions Using Small-Angle X-ray/Neutron Scattering.
- Anisotropic coarse-grain Monte Carlo simulations of lysozyme, lactoferrin, and NISTmAb by precomputing atomistic models.
- Flow Activation Energy of High-Concentration Monoclonal Antibody Solutions and Protein-Protein Interactions Influenced by NaCl and Sucrose.
- Morphological Characterization of Self-Amplifying mRNA Lipid Nanoparticles.
- A round-robin approach provides a detailed assessment of biomolecular small-angle scattering data reproducibility and yields consensus curves for benchmarking.
- Structural and biophysical properties of farnesylated KRas interacting with the chaperone SmgGDS-558.
- Conformational Heterogeneity of UCAAUC RNA Oligonucleotide from Molecular Dynamics Simulations, SAXS, and NMR experiments.
- Structural Characterization and Modeling of a Respiratory Syncytial Virus Fusion Glycoprotein Nanoparticle Vaccine in Solution.
- Chemical shifts-based similarity restraints improve accuracy of RNA structures determined via NMR.
- HIV-1 gp120-CD4-Induced Antibody Complex Elicits CD4 Binding Site-Specific Antibody Response in Mice.
- Structure of the cell-binding component of the Clostridium difficile binary toxin reveals a di-heptamer macromolecular assembly.
- Accuracy of MD solvent models in RNA structure refinement assessed via liquid-crystal NMR and spin relaxation data.
- Maximizing accuracy of RNA structure in refinement against residual dipolar couplings.
- Structural and Dynamical Order of a Disordered Protein: Molecular Insights into Conformational Switching of PAGE4 at the Systems Level.
- Comment on "Innovative scattering analysis shows that hydrophobic disordered proteins are expanded in water".
- PAGE4 and Conformational Switching: Insights from Molecular Dynamics Simulations and Implications for Prostate Cancer.
- A trapped human PPM1A-phosphopeptide complex reveals structural features critical for regulation of PPM protein phosphatase activity.
- Hybrid Applications of Solution Scattering to Aid Structural Biology.
- Prediction of nearest neighbor effects on backbone torsion angles and NMR scalar coupling constants in disordered proteins.
- Phosphorylation-induced conformational dynamics in an intrinsically disordered protein and potential role in phenotypic heterogeneity.
- Probing the Action of Chemical Denaturant on an Intrinsically Disordered Protein by Simulation and Experiment.
- Consistent View of Polypeptide Chain Expansion in Chemical Denaturants from Multiple Experimental Methods.
- Quantitative Characterization of Configurational Space Sampled by HIV-1 Nucleocapsid Using Solution NMR, X-ray Scattering and Protein Engineering.
- Side Chain Conformational Distributions of a Small Protein Derived from Model-Free Analysis of a Large Set of Residual Dipolar Couplings.
- Dynamic equilibrium between closed and partially closed states of the bacterial Enzyme I unveiled by solution NMR and X-ray scattering.
- Dissociation of glucocerebrosidase dimer in solution by its co-factor, saposin C.
- Quantitative residue-specific protein backbone torsion angle dynamics from concerted measurement of 3J couplings.
- Large interdomain rearrangement triggered by suppression of micro- to millisecond dynamics in bacterial Enzyme I.
- High accuracy of Karplus equations for relating three-bond J couplings to protein backbone torsion angles.
- Structural basis of hAT transposon end recognition by Hermes, an octameric DNA transposase from Musca domestica.
- Improved cross validation of a static ubiquitin structure derived from high precision residual dipolar couplings measured in a drug-based liquid crystalline phase.
- Dissociation of the trimeric gp41 ectodomain at the lipid-water interface suggests an active role in HIV-1 Env-mediated membrane fusion.
- Structure and dynamics of full-length HIV-1 capsid protein in solution.
- Crystal structures of the LsrR proteins complexed with phospho-AI-2 and two signal-interrupting analogues reveal distinct mechanisms for ligand recognition.
- Internal dynamics of the homotrimeric HIV-1 viral coat protein gp41 on multiple time scales.
- Sample preparation, data collection, and preliminary data analysis in biomolecular solution X-ray scattering.
- Contrast-matched small-angle X-ray scattering from a heavy-atom-labeled protein in structure determination: application to a lead-substituted calmodulin-peptide complex.
- Imino hydrogen positions in nucleic acids from density functional theory validated by NMR residual dipolar couplings.
- Monomeric α-synuclein binds Congo Red micelles in a disordered manner.
- Combined use of residual dipolar couplings and solution X-ray scattering to rapidly probe rigid-body conformational transitions in a non-phosphorylatable active-site mutant of the 128 kDa enzyme I dimer.
- Improved fitting of solution X-ray scattering data to macromolecular structures and structural ensembles by explicit water modeling.
- Solution structure of the 128 kDa enzyme I dimer from Escherichia coli and its 146 kDa complex with HPr using residual dipolar couplings and small- and wide-angle X-ray scattering.
- The impact of hydrogen bonding on amide 1H chemical shift anisotropy studied by cross-correlated relaxation and liquid crystal NMR spectroscopy.
- Structure/function implications in a dynamic complex of the intrinsically disordered Sic1 with the Cdc4 subunit of an SCF ubiquitin ligase.
- Site-specific backbone amide (15)N chemical shift anisotropy tensors in a small protein from liquid crystal and cross-correlated relaxation measurements.
- Solution structure and functional characterization of human plasminogen kringle 5.
- Chemical shift anisotropy of imino 15N nuclei in Watson-Crick base pairs from magic angle spinning liquid crystal NMR and nuclear spin relaxation.
- Using the experimentally determined components of the overall rotational diffusion tensor to restrain molecular shape and size in NMR structure determination of globular proteins and protein-protein complexes.
- NMR solution structure of the neurotrypsin Kringle domain.
- Solution structure of tRNAVal from refinement of homology model against residual dipolar coupling and SAXS data.
- Characterization of the solution structure of a neuroligin/beta-neurexin complex.
- The periplasmic domain of TolR from Haemophilus influenzae forms a dimer with a large hydrophobic groove: NMR solution structure and comparison to SAXS data.
- Refined solution structure of the 82-kDa enzyme malate synthase G from joint NMR and synchrotron SAXS restraints.
- Magnetic field induced residual dipolar couplings of imino groups in nucleic acids from measurements at a single magnetic field.
- Limits on variations in protein backbone dynamics from precise measurements of scalar couplings.
- Synaptic arrangement of the neuroligin/beta-neurexin complex revealed by X-ray and neutron scattering.
- [Thyroid cancer in male patients].
- Chemical shift tensors of protonated base carbons in helical RNA and DNA from NMR relaxation and liquid crystal measurements.
- Variable dimerization of the Ly49A natural killer cell receptor results in differential engagement of its MHC class I ligand.
- Pseudo-CSA restraints for NMR refinement of nucleic acid structure.
- Carbon-13 chemical shift anisotropy in DNA bases from field dependence of solution NMR relaxation rates.
- Refinement of multidomain protein structures by combination of solution small-angle X-ray scattering and NMR data.
- Weak alignment NMR: a hawk-eyed view of biomolecular structure.
- ABACUS, a direct method for protein NMR structure computation via assembly of fragments.
- Measurement of ribose carbon chemical shift tensors for A-form RNA by liquid crystal NMR spectroscopy.
- Protein structure elucidation from minimal NMR data: the CLOUDS approach.